

For most women and their families, the news that they have a baby with a chromosomal abnormality is perceived as bad – very bad! For all but the well informed, their initial image is of a physically deformed and mentally abnormal child, the antithesis of the ideal newborn, healthy and full of potential.
While that view may be justified for the majority of those with a chromosomal aneuploidy, it does not necessarily follow for individuals with a sex chromosome aneuploidy (SCA).
What does it mean to have a sex chromosome aneuploidy?
Complete or partial loss of sex chromosome – Turner’s syndrome
Turner’s syndrome (TS) is the result of total or partial monosomy of the X chromosome and is the most common SCA in female conceptions (affecting approximately three per cent). However, only one in 1000 embryos with a 45XO karyotype survives to term. The majority of affected pregnancies end in early miscarriage. The condition is primarily characterised by short stature and gonadal failure and frequently results in absent or arrested puberty. The spectrum of associated abnormalities include lymphedema, renal abnormalities, left-sided cardiac defects, hearing loss, various ocular abnormalities, and a number of distinctive phenotypic features such as webbed neck, low posterior hairline, low set ears, multiple nevi, nail dysplasia, short metatarsal and cubitus valgus. Intelligence falls well within the normal spectrum. However, some specific learning difficulties such as problems with spatial orientation and difficulties with social adjustment have been identified. A spectrum of health issues are more prevalent in women with TS. These include hypertension, celiac disease and endocrine diseases, particularly thyroid disease. In adult life, women with TS have an increased risk of cardiovascular morbidity and mortality. Aortic dissection or rupture appears to be an increasingly recognised cause of death in TS. This seems to be a particular risk for TS women in pregnancy.1
Individuals with a mosaic karyotype are less likely to have physical abnormalities or the characteristic phenotypic features of TS and are more likely to have preservation of ovarian function into puberty or adulthood. Consequently, many TS mosaics will go undiagnosed.
Study of Turner’s syndrome associated with partial monosomy has contributed to a better understanding of the cause of short stature in TS. Current evidence suggests that the short stature homobox-containing gene (SHOX) is located on the tip of the short arm of the X and Y chromosome.2 A rarer form of TS associated with deletion of the Y chromosome has helped with identification of the testis determining gene (SRY). Despite initial hope that progress in gene mapping may provide a clear relationship between karotypic abnormalities and clinical findings in TS, studies to date have been disappointing and short stature is the only clinical finding invariably associated with TS.
Early diagnosis and treatments, which include growth hormones to increase height potential3 and timely use of estrogen therapy to mimic normal puberty4, optimise outcome for TS individuals. Modern infertility treatment, particularly use of donor eggs, has assisted women with TS to achieve pregnancy. In the main, women with TS live happy and productive lives, albeit with a somewhat higher burden of health issues than women with 46XX karyotype and with an approximately doubled overall mortality ratio.
Extra sex chromosome syndromes
Klinefelter’s syndrome
Klinefelter’s syndrome (KS), 47XXY, results from the nondysjunction of the sex chromosomes and is associated with advancing parental age. It is therefore over-represented in cytogenetic abnormalities detected when screening for Down syndrome (DS). KS males are phenotypically normal at birth. The features associated with KS become increasingly apparent during adolescence and adulthood, and are characterised by tall stature, small testis and infertility on the basis of azoospermia. Overall, KS males’ IQ sits about ten points below their siblings’ scores. In the past, KS males were reported to have increased aggressive behaviour and tendency for criminal activity, but more careful research has not substantiated this association. It is likely that unless males with KS present for investigation of infertility, they will escape clinical diagnosis.
47XYY males
These individuals are tall and may have mild motor and language delay, but are otherwise normal with normal fertility.
47XXX (previously known as Triple X)
These individuals also tend to be tall and with normal fertility, but with some impairment of verbal and performance IQ at about 15 to 20 points below their siblings.
Other numerical sex chromosome abnormalities
Overall, there appears to be a relationship between the number of additional sex chromosomes and the severity of the phenotype. Moderate to severe intellectual handicap are usual.
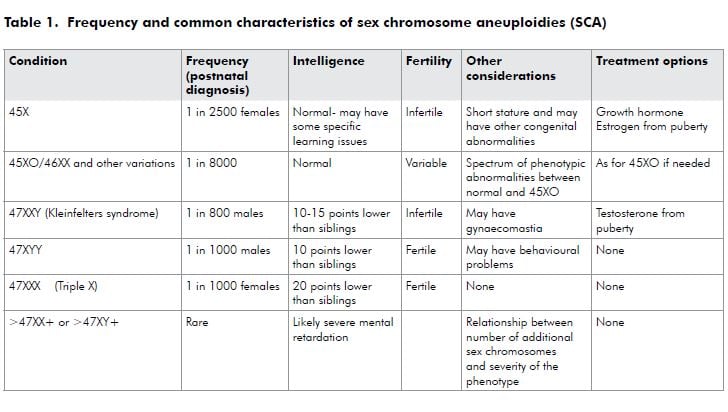
The types, frequency and common characteristics of SCAs are summarised below in Table 1.
What if SCA is diagnosed antenatally?
Prenatal diagnosis of SCA is likely to occur in two ways. Firstly and most commonly, the woman is diagnosed by chance after amniocentesis or chronic villi sampling (CVS) to exclude Down syndrome. SCAs represent approximately ten per cent of abnormal chromosome results identified following prenatal cytogenetics. Secondly, cytogenetic diagnosis of SCA may occur after investigation of an ultrasound abnormality.
Most individuals with a sex chromosomal abnormality are not identified because they do not present with a health issue which results in karyotype testing. The incidence of historically quoted abnormalities is likely to be an overestimate, where the denominator is TS individuals with abnormalities sufficient to result in karyotype. There is limited prospective follow-up information about individuals with a prenatal SCA diagnosis and preliminary data suggests that their prognosis is significantly more optimistic than previously appreciated. A recent study to evaluate the importance of this ascertainment bias in Turner’s syndrome has provided important information.5 Mosaic karyotype is the most common TS finding at prenatal diagnosis (56 per cent), whereas 45XO is the most common karyotype when diagnosis is made postnatally on the basis of phenotypic appearance or abnormalities (74 per cent). Further, the traditionally diagnosed group has almost twice as many phenotypic and cardiovascular abnormalities compared to the prenatally diagnosed group of individuals. These findings add weight to the impression that prenatally diagnosed TS is generally a ‘milder’ form of the disease. This is more likely to hold true for cases of SCA where diagnosis follows evidence of an ultrasound abnormality.
What to say?
Our responsibility as health professionals is to provide accurate context to the diagnosis of SCA. We must assist women to overcome their initial shock and preconceptions, and make a decision on whether to continue their pregnancy or seek a termination of pregnancy based on best information. The first communication with women is vital and affects how later information is interpreted, or even whether further information is sought. Evidence suggests that women with SCA are often provided with information in a haphazard manner by healthcare professionals.6
When faced with a diagnosis of SCA, the majority of women chose to terminate their pregnancy because of fears about having an ‘abnormal’ child. More recent research has shown that formal genetic counselling providing current and accurate information to women increases the likelihood that they will continue the pregnancy. Factors influencing parental decision-making is summarised in Table 2. How information is conveyed to women carrying children with an SCA affects both their decision to continue or terminate the pregnancy, and the degree of
anxiety they experience if they decide to continue.7 It is therefore important that obstetric units offering cytogenetic diagnosis have established protocols for sharing cytogenetic results with women, and for all staff who communicate these results to have accurate and up-to-date information about the conditions identified. This is particularly important when the diagnosis involves an SCA, as most obstetricians and midwives providing direct care will have very limited personal experience to draw on. For women deciding to continue their pregnancies, community organisations such as the Turner’s Syndrome Society, which has branches in Australia and New Zealand, may provide helpful ongoing support.

References
- Karnis MF, Zimon AE, Lalwani SI, et al. Risk of death in pregnancy achieved through oocyte donation in patients with Turner Syndrome: a national survey. Fert Steril. 2003; 80:498-501.
- Ross JL, Scott C Jr, Marttila P, Kowal K. Phenotypes associated with SOX Deficiency. J Clin Endocrinol Metab. 2001; 86;5674-5680.
- Sas TC, de Muinck Keizer-Schrama,SM, Stijen T, et al. Normalisation of height in girls with Turner Syndrome after long-term growth hormone treatment: results of a randomised dose response trial. J Clin Endocrinol Metab. 1999; 84:4607-4612.
- Donaldson MD, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer. Arch Dis Child. 2006; 91(6):513-20.
- Daniel FG, Eugster E, Zager AJ, et al. Ascertainment bias in Turner Syndrome: New insights from girls who were diagnosed incidentally in prenatal life. Pediatrics 2004; 114: 640-644.
- Abramsky L, Hall S, Levitian J, et al. What parents are told after prenatal diagnosis of a sex chromosome abnormality: interview and questionnaire study. BMJ 2001; 322: 463-466.
- Shaw SW, Chueh HY, Chang SD, et al. Perental decision regarding prenatally detected fetal chromosomal abnormality and the impact of genetic counselling. An analysis of 57 cases in Taiwan. Aust & NZ J of O&G 2008; 48: 155-159.



Leave a Reply