

Vaccination has been successfully implemented for the control of infectious diseases for more than 200 years. Since Edward Jenner (see Figure 1) first developed the smallpox vaccine in 1798, seminal work by many other scientists has led to the development of a number of different vaccines, targeted against various disease-causing viruses and bacteria. The success of immunisation cannot be denied: the mortality rate of diseases for which vaccines are available has decreased by more than 97 per cent and, in some cases, has led to the complete eradication of the disease (for example, smallpox, which was eradicated in 1980).1
Aligning with the adage ‘prevention is better than cure’, the basic premise behind vaccination is the controlled introduction of a non-infectious form of a target pathogen into the body, to allow development of specific immunity against the virus or bacterium, without causing a severe infection. While this process is relatively well understood by medical practitioners, the science behind how new vaccines are designed and developed into safe formulations may be less familiar. This article will explore how different viral vaccines are made and outline the complex journey involved in getting a new vaccine from the laboratory bench to the patient.
Viruses and the immune response
Viruses are incredibly small, highly infectious organisms that can only survive and replicate inside the living cells of a host organism (such as humans). The structure of a virus is relatively simple, typically consisting of two main entities: genetic material (either DNA or RNA) and a protective coating surrounding the genetic material, mainly consisting of proteins.2 This protein shell contains a number of different and unique peptide segments, referred to as antigens, that the body uses to identify the virus. For example, in much the same way that humans generally distinguish each other through differences in hair and skin colours, the human body can identify and distinguish foreign organisms using these antigens.

Figure 1. Edward Jenner, whose pioneering observation that cowpox gave immunity to smallpox led to the science of vaccination.
Once a virus is inside a host cell, it can grow and proliferate at incredibly high speeds, hijacking cell after cell and rapidly causing an infection. Luckily for us, the body is programmed to respond to the presence of foreign pathogens quickly, via activation of the innate immune response.3 This causes the release of various molecules, which attack the virus and helps to protect the body during the initial infection. Following this, the adaptive immune response is then activated. It consists of the cell-mediated (T-cell) and humoral (B-cell) immune responses that work in partnership to handle much of the dirty work that is involved during elimination of a virus from the host. A key event during the humoral immune response is the production of antibodies (immunoglobulins) that recognise and bind to the specific antigens of a virus, blocking the virus-host cell interactions or lysing virus-infected cells.4 5 The production of memory B-cells is also activated during this response, which leads to long-lasting immunity and a faster initial immune response upon re-infection of the host with the same virus.
Table 1. A list of the different viral pathogens that humans can be immunised against, categorised according to their vaccine type.6 7
| Vaccine type | Viral pathogen |
| Live, attenuated | Adenovirus
Influenza (nasal spray) Measles Mumps Poliovirus (OPV) Rotavirus Rubella Smallpox Varicella (chickenpox) Yellow fever Zoster (shingles) |
| Whole inactivated/ killed | Hepatitis A Influenza (injection) Japanese encephalitis Poliovirus (IPV) Rabies |
| Recombinant/ purified subunit | Hepatitis B
Human papillomavirus Influenza (injection) |
Isolate, inactivate, inject
To give the body a head start in developing immunity against these viruses, early development of vaccines was focused on Louis Pasteur’s principles of ‘isolate, inactivate, inject’ the causative organism.8 9 In a nutshell, this involved isolating mutated strains or inducing a mutation in the virulent (wild-type) form of the virus to generate a strain with reduced or eliminated ability to replicate within the host. Termed ‘live, attenuated’ (see Table 1), these vaccines are highly effective at eliciting an immune response in the body, as they imitate the native, non- attenuated virus in nearly every way.10 11 Some examples of these vaccines include chickenpox, shingles and yellow fever.
A second reason why many of these live, attenuated viruses make particularly effective vaccines is because the antigens produced by the virus do not change very much, meaning that humans can be immunised for life against many of these pathogens. One risk associated with these vaccines though is spontaneous reversion of the attenuated strain of the virus back to the wild-type, potentially causing disease in patients who have received the vaccine.Many cases of reversion have been reported for the oral poliovirus vaccine (OPV) that immunises humans against all three poliovirus types. Thus, given its associated risks, a different poliovirus vaccine – the inactivated poliovirus vaccine (IPV) – was developed and is now used preferentially to immunise against poliovirus, even though its protective action is not as good as OPV.12 13
In light of the above example, a second class of vaccines was developed for those pathogens where immunising with the attenuated virus is not feasible or not possible.14 Therefore, rather than attenuating the virus to make it less virulent, some viruses are wholly inactivated or killed to make the vaccine. This is usually achieved by treating the live organism with chemical agents, such as formaldehyde, which renders the virus unable to penetrate cells or replicate in vivo.15 As a result of this, whole inactivated or killed vaccines are generally less immunogenic than live, attenuated vaccines owing to their inherent loss of function. For this reason, a compound called an adjuvant is usually added to the vaccine formulation to help stimulate the immune system and promote longer lasting immunity. Adjuvants are typically aluminium salts or oil-in-water emulsions that help to increase or modulate the immune response.16 Some examples of whole inactivated or killed vaccines are those for hepatitis A and Japanese encephalitis.17 18
In addition to inactivating or killing the virus, a third method for making vaccines involves isolating the purified protein of the virus shell, thus eliminating the viral genetic material from the vaccine formulation. This ‘recombinant’ process has been successfully implemented for the preparation of the hepatitis B vaccine.19 20 Briefly, the basic laboratory process involves inserting the genetic code for the hepatitis B surface antigen HBsAg into yeast cells, which are then grown, before the protein is isolated from the cells, purified and formulated into the vaccine.21
New tricks, new vaccines
More recently, new methodologies have been developed for vaccine production against pathogens exhibiting high antigenic variability, such as the human papillomavirus (HPV). HPV has more than 170 different strains, many of them causing diseases such as cervical and oropharyngeal cancers, and genital warts.22 Therefore, in order to be broadly protective, any HPV vaccine must incorporate many of these strains. Early development of the HPV vaccine began via expression and purification of the individual proteins (L1 and L2) of the virus shell of a cancer- causing HPV strain (HPV16) in an attempt to replicate the outer shell of the native virus.23 24 While these proteins could be grown from cells in the lab, it was only under the right conditions that the L1 and L2 proteins self-assembled to form virus-like particles (VLPs) that mimicked the native shell of the virus. In this seminal work led by Prof Ian Frazer and Dr Jian Zhou, this procedure turned out to be the key trick in the development of the human papillomavirus vaccine as it then allowed for the preparation of a number of VLPs for many of the different HPV strains.25 26 Thus, incorporating four different VLPs from the HPV types 6, 11, 16 and 18, the quadrivalent HPV vaccine Gardasil (Merck) now confers protection against the HPV strains that are responsible for 70 per cent of cervical cancers in women and 90 per cent of genital warts cases.
Table 2. The different phases of clinical trials required for approval to license vaccines.27
| Clinical trial stage | Number of subjects tested | Tests conducted |
| Phase I | <100 | Safety Immunogenicity |
| Phase II | >100–1000 | Safety Immunogenicity Dosing Scheduling Method of delivery |
| Phase III | 1000 to >10 000 | Safety Efficacy |
Trepidation and trials
While the discovery and initial development phase of a vaccine can take many years, the process involved in taking a vaccine from the bench to the patient is very lengthy and costly. During the discovery phase of a vaccine, laboratory and animal tests are conducted as the recommended pre-clinical trials. If the vaccine passes all of these tests, it then enters into the various phases for testing on humans, as shown in Table 2. Given the rigorous nature of these tests, many vaccine candidates often fail in either Phase I or Phase II trials. If a vaccine candidate is deemed efficacious and safe, what then follows is the approval and licensure of the vaccine, after which it is then made publicly available.28 The time that this whole process takes does vary depending on each vaccine, however to give an example – in the most recent case of the HPV vaccine, the process from making the first HPV16 virus-like particle in the laboratory to the first licensed human immunisation took a total of 14 years.29
Vanguard vaccine development
While we can celebrate past successes in the realm of vaccination, we must also look forward to the development of new vaccines against a number of different disease-causing viruses for which no preventative treatment exists, including human immunodeficiency virus (see Figure 2), ebola and malaria.30 Thankfully, science is providing important steps towards the development of new vaccines using a few different approaches. At the forefront of these technologies is ‘reverse vaccinology’, a process that involves sequencing the genome of a particular pathogen to identify antigenic sequences that can be used in the development of new vaccines. The key benefit of this process is that the vaccine is targeted only towards the antigens of interest and therefore the immune response is highly specific towards the target virus.31
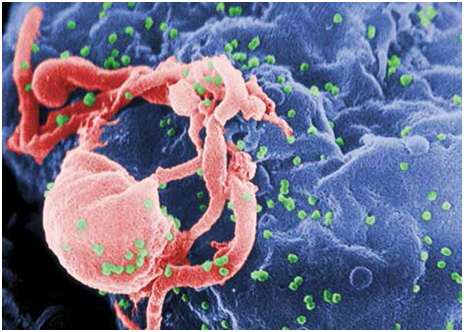
Figure 2. Scanning electron micrograph of HIV-1 budding (in green) from cultured lymphocyte. This image has been coloured to highlight important features. Multiple round bumps on cell surface represent sites of assembly and budding of virions.
Structural vaccinology is another technology that is being used for vaccine development against viruses with high antigenic variability. Briefly, this process looks more closely at the structure and prominence of different antigenic sequences within a viral protein and uses this information to guide the engineering of a new vaccine candidate, particularly when key antigens are obscured from the body’s immune system.32 Other methods of developing vaccines include instructive immunotherapy or programming of the immune system; and use of ‘nano-carriers’ for antigen delivery, such as liposomes, nanoparticles and dendrimers.33 34
While there are still a number of disease-causing pathogens with high rates of morbidity and mortality; the past 200 years of vaccine development has provided many success stories that we can be proud of. In particular, the pioneering work of Prof Ian Frazer and Dr Jian Zhou on the development of the virus-like particle and subsequent HPV vaccine is a recent example. In light of this, we can be confident that in the future many of these viruses will be outsmarted by next generation vaccines currently sitting at the forefront scientific investigation.
References
- Plotkin SA, editor. History of Vaccine Development. New York: Springer; 2011.
- Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 19-63.
- Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 19-63.
- Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 19-63.
- Chaplin DD. Overview of the human immune response. J Allergy Clin Immunol.2006;117:S430-S435.
- Vaccines & Immunizations [Internet]. Georgia: National Center for Immunization and Respiratory Diseases; c20001 [updated 2015 Feb 23; cited 2015 Feb 25]. Available from: www.cdc.gov/vaccines/.
- Ulmer JB, Valley U, Rappuoli R. Vaccine manufacturing: challenges and solutions. Nat Biotechnol. 2006;24(11):1377-1383.
- Plotkin SA, editor. History of Vaccine Development. New York: Springer; 2011.
- Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotech. 2007;25(12):1361-1366.
- Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 19-63.
- Chaplin DD. Overview of the human immune response. J Allergy Clin Immunol.2006;117:S430-S435.
- Plotkin SA, editor. History of Vaccine Development. New York: Springer; 2011.
- Vaccines & Immunizations [Internet]. Georgia: National Center for Immunization and Respiratory Diseases; c20001 [updated 2015 Feb 23; cited 2015 Feb 25]. Available from: www.cdc.gov/vaccines/.
- Bagnoli F, Baudner B, Mishra RPN, Bertolini E, Fianschi L, Mariotti P, Nardi-Dei V, Boucher P, Rappuoli R. Designing the Next Generation of Vaccines for Global Public Health. OMICS A J Integrat Biol.2011;15(9):545-566.
- Bagnoli F, Baudner B, Mishra RPN, Bertolini E, Fianschi L, Mariotti P, Nardi-Dei V, Boucher P, Rappuoli R. Designing the Next Generation of Vaccines for Global Public Health. OMICS A J Integrat Biol. 2011;15(9):545-566.
- O’Hagan DT, Gregoria ED. The path to a successful vaccine adjuvant – ‘The long and winding road’. Drug Discovery Today. 2009
Jun;14(11/12):541-551. - Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 19-63.
- Ulmer JB, Valley U, Rappuoli R. Vaccine manufacturing: challenges and solutions. Nat Biotechnol. 2006;24(11):1377-1383.
- Lipowsky G, Bachmann MF. Design and Development of Recombinant Vaccines with Viral Properties. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.;2011; pp 19-63.
- Ulmer JB, Valley U, Rappuoli R. Vaccine manufacturing: challenges and solutions. Nat Biotechnol. 2006;24(11):1377-1383.
- Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotech. 2007;25(12):1361-1366.
- Bzhalava D, Guan P, Franceschi S, Dillner J, Clifford G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology. 2013 Jul 16;445:224-231.
- Zhou J, Sun XY, Stenzel DJ, Frazer IH.Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology. 1991 Nov;185(1):251-257.
- King M. Ian Frazer: The man who saved a million lives. Brisbane, Australia: University of Queensland Press; 2013.
- Zhou J, Sun XY, Stenzel DJ, Frazer IH.Expression of vaccinia recombinant HPV 16 L1 and L2 ORF proteins in epithelial cells is sufficient for assembly of HPV virion-like particles. Virology. 1991 Nov;185(1):251-257.
- King M. Ian Frazer: The man who saved a million lives. Brisbane, Australia: University of Queensland Press; 2013.
- Vaccines, Blood & Biologics [Internet].Maryland: U.S. Food and Drug Administration (FDA); c2009 [updated 2009 Jun 18; cited 2015 Feb 25]. Available from: www.fda.gov/biologicsbloodvaccines/developmentapprovalprocess/biologicslicenseapplicationsblaprocess/ucm133096.htm .
- Vaccines, Blood & Biologics [Internet]. Maryland: U.S. Food and Drug Administration (FDA); c2009 [updated 2009 Jun 18; cited 2015 Feb 25]. Available from: www.fda.gov/biologicsbloodvaccines/ developmentapprovalprocess/ biologicslicenseapplicationsblaprocess/ ucm133096.htm .
- Vaccines, Blood & Biologics [Internet]. Maryland: U.S. Food and Drug Administration (FDA); c2009 [updated 2009 Jun 18; cited 2015 Feb 25]. Available from: www.fda.gov/biologicsbloodvaccines/ developmentapprovalprocess/biologicslicenseapplicationsblaprocess/ucm133096.htm.
- Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotech. 2007;25(12):1361-1366.
- Rosini R, Barocchi MA, Rappuoli R. Microbial Vaccine Design: The Reverse Vaccinology Approach. In Development of Vaccines. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011; pp 1-18.
- Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotech.2007;25(12):1361-1366.
- Rappuoli R. Bridging the knowledge gaps in vaccine design. Nat Biotech. 2007;25(12):1361-1366.
- D’Souza MJ, Tawde SA, Akalkotkar A, Chablani L, D’Souza M, Chiriva-Internati M. Nanotechnology in Vaccine Delivery. In Geise M, editor. Molecular Vaccine. Vol 2. Switzerland: Springer International Publishing; 2014; pp 727-741.



Leave a Reply