

Menarche is an important milestone of pubertal transition and has been widely viewed as a sign of physical and emotional health.1 Timely onset of menstrual flow indicates functioning neuroendocrine-reproductive system and often reassures that other physiological changes of puberty progress normally. Unsurprisingly, teenage girls and their guardians tend to get worried about delayed onset of menses.
Primary amenorrhea is defined as a failure to reach menarche. The estimated incidence is 0.1–0.3 per cent and is far less common than that of secondary amenorrhoea (3–4 per cent).2
When to evaluate?
Menarche usually occurs at around 12–13 years of age, within three years of breast development when most girls have Tanner breast stage IV. By age 15, up to 98 per cent will have had menses.3 4 Evaluation is indicated when any of the following criteria are met:5 6 7
- Absence of menarche by age 15 years in the presence of normal secondary sexual characteristics (2.5 standard deviations (SD) above the mean of 13 years)
- Menarche has not occurred within three years of breast development
- Menarche has not started by age 14 years in the presence of hirsutism, excessive exercise or clinical suspicion of eating disorder or outflow tract obstruction
- No breast development by age 13 years (2.5 SD above the mean of 10 years)
Assessment
Most causes of primary and secondary amenorrhoea are similar and both conditions should be approached in the same way.8 All evaluations should begin with exclusion of pregnancy, regardless of the sexual history.
History should focus on chronology of pubertal changes, eating and exercise patterns, weight change, chronic illness, medication use, sexual activity and symptoms of galactorrhea, thyroid dysfunction and androgen excess. Family history should include age at menarche and menopause, genetic disorders and developmental delay. Physical examination includes assessment of habitus, breast development, hirsutism, Turner stigmata, thyroid palpation and examination of genitalia.
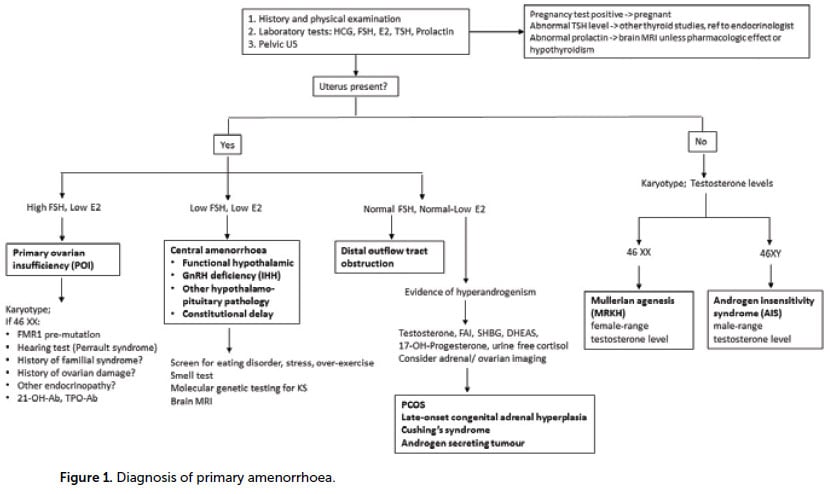
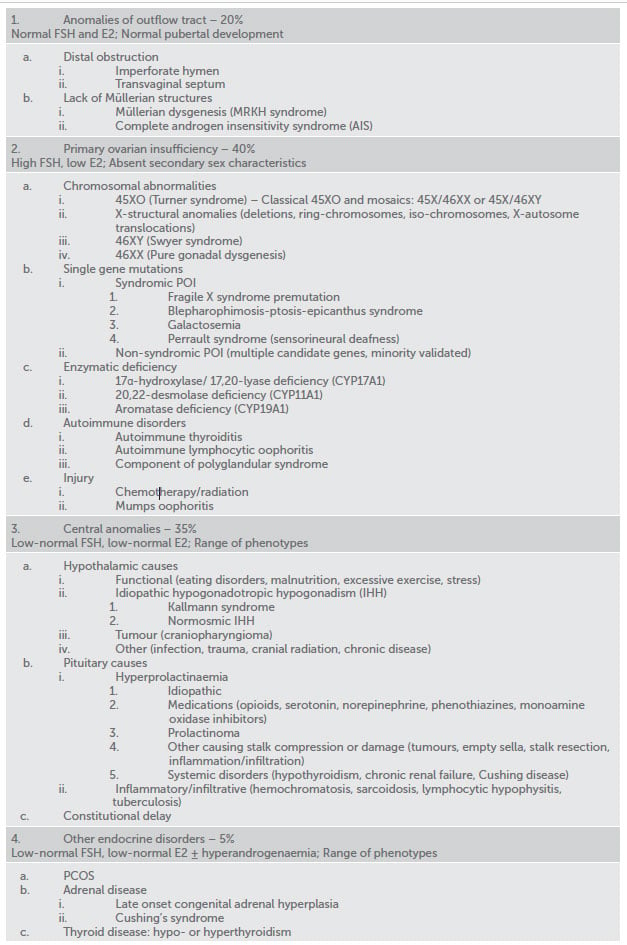
Table 1. Common causes of primary amenorrhoea.
The initial evaluations include transabdominal ultrasound and serum FSH, E2, TSH and prolactin. Subsequent investigations largely depend on presence of the uterus and FSH/E2 levels (Figure 1).
Causes of primary amenorrhoea
Amenorrhoea may result from an abnormality at any level of the reproductive tract and there is a long list of potential causes (Table 1). It is useful to think about four broad categories:
- Anomalies of the outflow tract
- Primary ovarian insufficiency
- Central anomalies (hypothalamic-pituitary)
- Other endocrine disorders
Anomalies of the outflow tract
Anatomical defects represent 20 per cent of the causes of primary amenorrhoea and are manifested by normal pubertal development. Distal obstruction (imperforate hymen and transverse vaginal septum) commonly presents with cyclic pelvic pain due to hematocolpos. The diagnosis is made on examination. Surgical resection is a definite treatment and fertility is not compromised.
Lack of Müllerian structures is another group of outflow tract anomalies, which includes Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH) and androgen insensitivity syndrome (AIS). MRKH (1:4500 women) is a multifactorial genetic syndrome, featured by vaginal agenesis and uterine maldevelopment from a rudimentary to an absent uterus. Associated skeletal, renal and auditory anomalies are common.9 Diagnosis is possible with ultrasound or MRI. AIS (1:20,400 newborn males) is caused by end-organ insensitivity to androgens in genetical males with functioning testes. Both conditions share common clinical features; the differential diagnosis is made by karyotype. Management includes psychosocial counselling, creation of neovagina, removal of uterine remnants with active endometrium (MRKH) or gonadectomy due to risk of malignancy in undescended testes (AIS). Fertility interventions include surrogacy (MPKH) and surrogacy plus oocyte donation (AIS).
Primary ovarian insufficiency
Primary ovarian insufficiency (POI) or hypergonadotropic hypogonadism is characterised by gonadal dysfunction due to abnormal migration or rapid depletion of germ cells. Main features are high FSH, low E2 and abnormal pubertal development.
Chromosomal derangements are a well-recognised cause of POI in adolescents, of which Turner syndrome is the most common (1:2000–1:4000 live born girls). Classical monosomy (45XO) accounts for 50–60 per cent of karyotypes and is commonly diagnosed before puberty due to distinctive characteristics (short stature, webbed neck, low hairline and shield chest). Turner mosaics have broad phenotypic variations and are more likely to be discovered during evaluation of amenorrhoea.10 Common health issues include cardiac and kidney anomalies, autoimmune disorders (diabetes and thyroiditis), obesity, strabismus and cataract. 46XY (Swyer syndrome) is featured by non-functional gonads that don’t produce anti-Müllerian hormone or androgens. The affected individuals have normal female phenotype and intact Müllerian structures. Other chromosomal abnormalities and 46XX pure gonadal dysgenesis are far less common.
Multiple genes on X chromosome, autosomes and mitochondria have been proposed as candidate markers of POI, although only a few were functionally validated.11 The most common single-gene disorder linked with POI is Fragile X premutation, caused by a higher number (54–200) of CGG repeats on FMR1 gene. POI is not observed with full mutation. It occurs in 15–20 per cent of FMR1 premutation carriers after the age of 18 in association with secondary amenorrhoea, although the pubertal phenotypes in adolescent carriers are still poorly addressed.12
Uncommon presentations in adolescents include deficiencies in steroidogenic pathway enzymes, autoimmune conditions and iatrogenic causes related to surgery, chemo- and radiotherapy.
All individuals with non-iatrogenic POI should have chromosomal analysis. In patients with Turner syndrome, additional evaluations include echocardiogram, eye test, renal imaging, thyroid studies and diabetes screen. FMR1 premutation testing is considered if normal karyotype. Other genetic testing is not indicated, unless specific syndrome is suspected.13 There is no validated marker to confirm diagnosis of immune POI. Testing of 21OH-Ab (adreno-cortical antibodies) and thyroid TPO-Ab can be considered if the cause of POI is unclear or when immune condition is suspected.14
Induction of puberty in adolescents with gonadal failure includes low-dose oestrogen from age 12, with a gradual increase over 2–3 years. Cyclic progestogen is added two years later upon completion of breast development, to avoid misshapen tubular breasts. Adequate oestrogen-progestin replacement throughout the reproductive years is recommended to prevent osteoporosis, reduce risk of cardiovascular disease, normalise sexual function and reduce possible risk of cognitive impairment.15 The data to support androgen replacement is limited.16 Gonadectomy is recommended in individuals with Y chromosome due to 20–30 per cent risk of malignancy. Psychosocial support and monitoring of bone health are important. Oocyte donation remains the main option for fertility in these women.
Box 1. Summary
The key practice points in approach to primary amenorrhoea:
- Pregnancy should be excluded in all patients who present with amenorrhoea
- The initial investigations include FSH, TSH, prolactin and pelvic ultrasound
- Karyotype should be considered in individuals with absent uterus or POI
- In patients with 46XY, intra-abdominal gonads have malignant potential and need to be removed
- Combined OCP are contra-indicated for induction of puberty
- Patients with persistent hypoestrogenism are at risk for osteoporosis and should be screened with DEXA and treated with vitamin D, calcium and oestrogen therapy where appropriate
- In patients with functional hypothalamic amenorrhea, COCP does not improve bone density unless nutritional deficiencies are corrected
- Differentiation of hypothalamic amenorrhoea from PCOS depends on clinical judgement and presence of hyperandrogenism
Central amenorrhoea
Hypothalamic disorders are featured by hypogonadotropic hypogonadism due to abnormal secretion or inhibition of gonadotrophin-releasing hormone (GnRH).
Functional hypothalamic amenorrhea is common in adolescents and occurs when the hypothalamic-pituitary-ovarian axis is suppressed due to an energy deficit caused by weight loss, excessive exercise or stress.17 These patients should be screened for eating disorders and malabsorption syndromes (coeliac disease). Good nutrition, optimal body weight and stress reduction are primary treatment goals. Menses commonly restore at 90 per cent of ideal body weight. Bone density evaluation and calcium/vitamin D supplements are important. Combined oral contraceptive pills (COCP) restore menses, but don’t improve bone density without adequate nutritional rehabilitation.
More rare causes include congenital GnRH deficiencies, comprising a heterogeneous group of isolated hypogonadotropic hypogonadism (IHH). The condition may be sporadic, autosomal dominant or X-linked recessive disorder with varying phenotypes.18 Kallmann syndrome (1:120,000 females), a form of IHH featured by anosmia, is caused by abnormal migration of GnRH and olfactory neurons. The most common genes implicated in the disorder include ANOS1 (KAL1), CHD7, FGF8, FGFR1, PROK2, or PROKR2. Other, normosmic forms, are linked with mutations in genes responsible for GnRH production and secretion, although full molecular pathogenesis of the disorder is unclear.19 The diagnosis is clinical. Negative molecular genetic testing will not rule out a possible diagnosis, as many associated genes are still unknown. The management is induction of puberty and long-term oestrogen-progestin replacement. Ovulation induction with exogenous gonadotrophins is a first-line reproductive option.
Prolactin inhibits secretion of pituitary gonadotrophins and can be elevated because of medications, pituitary adenoma, hypothyroidism or pituitary stalk compression when inhibition by hypothalamic dopamine is compromised. Hyperprolactinaemia, unless related to primary hypothyroidism or medications, warrants MRI of pituitary. Dopamine agonists are the first-line management.20 Other disorders include tumours, empty sella syndrome, infection, trauma or autoimmune destruction of the pituitary, but these are relatively uncommon in adolescents.
Constitutional delay, when patients will have a later onset of normal puberty, is seen more commonly in boys. This is a diagnosis of exclusion and family history of late menarche is common.21
Other endocrine disorders
Polycystic ovary syndrome (PCOS) is a common endocrine condition affecting 12–21 per cent of reproductive-aged women.22 The Rotterdam diagnostic criteria require presence of two of the following: oligo-ovulation, clinical/biochemical hyperandrogenism or polycystic ovaries on ultrasound, after excluding other aetiologies. Importantly, polycystic ovarian morphology on ultrasound is not specific in adolescents and is not recommended criterion in this age group.23 While typically associated with secondary amenorrhoea, PCOS can manifest with primary amenorrhoea in adolescents with higher levels of circulating androgens. Cardiometabolic risk screening with OGTT and fasting lipids, along with assessment of blood pressure, BMI and waist circumference is recommended in all women with PCOS regardless of age. This should be repeated annually in overweight/obese individuals and every two years in those with normal BMI.24 Initial management includes lifestyle modifications, restoring menstrual cyclicity, managing symptoms of androgen excess and psychological support. COCP is commonly considered in women not planning pregnancy. Ovulation induction with clomiphene or letrozole is fertility treatment of choice in anovulatory women with PCOS.25
Androgen-producing tumour, late-onset congenital adrenal hyperplasia and Cushing’s syndrome are uncommon, but should be considered and distinguished from PCOS in the evaluation of hyperandrogenic amenorrhea. Both hypo- and hyperthyroidism cause menstrual abnormalities, while more severe forms may result in amenorrhoea. Timely referral to an endocrinologist and correction of underlying endocrinopathy is commonly sufficient for maintaining normal menstrual and reproductive function.
Conclusion
Amenorrhea is a symptom, not a disease. Depending on its cause, failure to attain menarche may lead to varying clinical sequalae. Timely evaluation and consideration of a broad differential diagnosis have important implications for emotional, physical and reproductive health in young women.
References
- Adams Hillard PJ. Menstruation in adolescents: what do we know? And what do we do with the information? J Pediatr Adolesc Gynecol. 2014;27:309-19.
- Timmreck LS, Reindollar RH. Contemporary issues in primary amenorrhea. Obstet Gynecol Clin North Am. 2003;30:287-302.
- Chumlea WC, Schubert CM, Roche AF, et al. Age at menarche and racial comparisons in US girls. Pediatrics. 2003;111:110-3.
- Warren D, Yu M. Growing Up in Australia: The Longitudinal Study of Australian Children (LSAC). Australian Institute of Family Studies. Pubertal status and emotional, school and social functioning. LSAC Annual Statistical Report 2015. Chapter 6:127-51.
- Diaz A, Laufer MR, Breech LL. American Academy of Pediatrics Committee on Adolescence; American College of Obstetricians and Gynecologists Committee on Adolescent Health Care, Menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Pediatrics. 2006;118:2245-50.
- American College of Obstetricians and Gynecologists. Committee Opinion No. 651. Menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Obstet Gynecol. 2015;126:e143-6.
- Practice Committee of American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2008;90(5 Suppl):S219-25.
- Practice Committee of American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2008;90(5 Suppl):S219-25.
- Choussein S, Nasioudis D, Schizas D, Economopoulos KP. Müllerian dysgenesis: a critical review of the literature. Arch Gynecol Obstet. 2017;295:1369-81.
- Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med. 2004;351(12):1227-38.
- Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21:787-808.
- De Caro J, Dominguez C, Sherman S. Reproductive health of adolescent girls who carry the FMR1 premutation: Expected phenotype based on current knowledge of fragile X-associated primary ovarian insufficiency. Ann NY Acad Sci. 2008;1135:99-111.
- ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31:926-37.
- ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31:926-37.
- ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31:926-37.
- ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31:926-37.
- Gordon CM. Clinical practice. Functional hypothalamic amenorrhea. N Engl J Med. 2010;363:365-71.
- Shaw ND, Seminara SB, Welt CK, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab. 2011;96:E566-76.
- Shaw ND, Seminara SB, Welt CK, et al. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab. 2011;96:E566-76.
- Practice Committee of American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril. 2008;90(5 Suppl):S219-25.
- De Luca F, Argente J, Cavallo L, et al. Management of puberty in constitutional delay of growth and puberty. J Pediatr Endocrinol Metab. 2011;14:953-7.
- Evidence-based guideline for the assessment and management of polycystic ovary syndrome. Jean Hailes for Women’s Health on behalf of the PCOS Australian Alliance. Melbourne, 2015.
- Evidence-based guideline for the assessment and management of polycystic ovary syndrome. Jean Hailes for Women’s Health on behalf of the PCOS Australian Alliance. Melbourne, 2015.
- Evidence-based guideline for the assessment and management of polycystic ovary syndrome. Jean Hailes for Women’s Health on behalf of the PCOS Australian Alliance. Melbourne, 2015.
- Evidence-based guideline for the assessment and management of polycystic ovary syndrome. Jean Hailes for Women’s Health on behalf of the PCOS Australian Alliance. Melbourne, 2015.



Leave a Reply