

Many health professionals are unsure what the term intersex means; it can be a confusing term for those who are not familiar with it. Intersex refers to variations in the development of sex characteristics that do not fit the typical norms of male or female. There may be a variation in the chromosomes, gonads or reproductive anatomy of the individual, and results in a blend of ‘typical male’ and ‘typical female’ characteristics.1 Understandably, this covers a diverse spectrum of variations and accounts for up to one to two per cent of the population.2
Medical texts refer to intersex variations as disorders or differences of sex development (DSD). While these terms are still commonly used within medical literature and the medical community, they have been challenged recently by intersex advocates as ‘othering’ and pathologising, with associated negative connotations.3 In response, some clinicians have rephrased DSD to use the term ‘diverse sex development’.
In addition, the term ‘intersex’ is distinct from the terms ‘transgender’ or ‘gender diverse’. Transgender people have an identity that is different from that of their assigned gender. To be transgender, there does not need to be any underlying variation in development of sex characteristics in the body. While some individuals who are intersex may have a gender identity different to their assigned gender, others may not.
History of nomenclature
Over time, the terminology used to describe intersex variations has changed and evolved. Western medicine tends to favour the use of labels and descriptors. Many years ago, the medical terminology used for people with intersex variations included ‘true hermaphrodite’, ‘pseudo hermaphrodite’, ‘testicular feminisation’ and ‘sex reversal’. In an attempt to update terminology, the ‘Chicago consensus statement on the management of intersex disorders’ was published in 2006.4 The statement, produced by a large group of professionals working in the field, rewrote the terminology with the aim of using more scientifically descriptive and less pejorative medical terms. Terminology included the ‘DSD’, as well as descriptive names such as ‘complete androgen insensitivity syndrome’. These are the terms now widely used in medical practice, medical literature and research.
Appropriate language
The terminology a person with variations of sex characteristics chooses to use is entirely individual and there is no ‘one size fits all’. Many intersex individuals do not prescribe to the phrase ‘DSD’, preferring the term ‘intersex’. However, other individuals may not use the term ‘intersex’ and prefer something different, such as their specific medical diagnosis. As health professionals, we must be aware of the sensitive nature of these terms and seek to use affirming language preferred by the individual concerned.
The reproductive developmental pathway
Until six weeks of development (eight weeks gestation), growth is the same for all embryos. This includes a bipotential gonad and the presence of both Müllerian and Wolffian ducts. The undifferentiated gonad typically develops into either a testis or an ovary, and the phenotypic sex develops depending on what hormones are produced by the gonad (Figure 1).
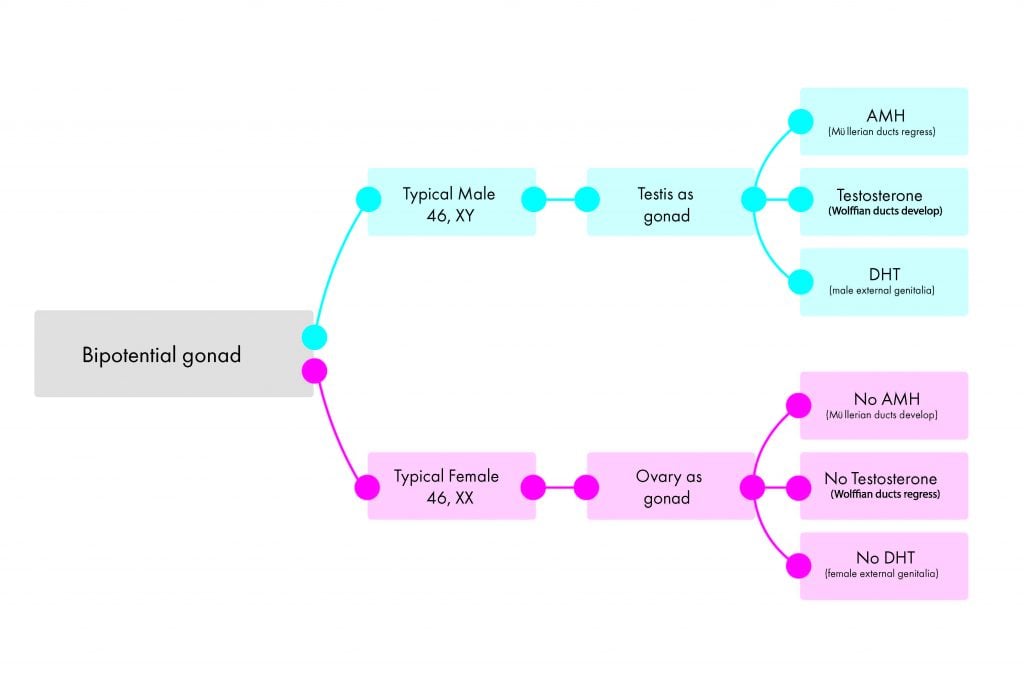
Figure 1. Summary of the typical development of the male and female reproductive systems.
In the typical male pathway, presence of the SRY gene, along with a complex interplay of other genes (for example, SOX9), leads to the development of the testes as the gonad. Anti-Müllerian hormone (AMH) produced by the testes causes regression of the Müllerian ducts. Production of testosterone promotes development of the Wolffian ducts to the internal male reproductive structures. Testosterone is peripherally converted to dihydrotestosterone (DHT) via the 5 alpha-reductase enzyme, and results in virilisation of the external genitalia, to give the appearance of ‘typical male’.
In the typical female pathway, the absence of the SRY gene, along with the active upregulation of other genes (for example, WNT4), promotes the development of the ovary as the gonad. Lack of AMH leads to the development of the Müllerian ducts to the internal female reproductive tract. Lack of locally produced testosterone leads to regression of the Wolffian ducts. Finally, lack of androgen exposure results in ‘typical female’ external genitalia (clitoris, urethra, lower vagina and labia) developing.
Variations in sex characteristics
Variations to the ‘typical male’ and ‘typical female’ pathway can occur at any level of development. As intersex variations cover a wide range of situations, it is impossible to describe them all here. Below outlines the developmental pathway for three more common examples of intersex variations.
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) occurs when the body has a deficiency in a specific enzyme required for converting hormones along the steroid hormone pathway. The most common enzyme deficiency in CAH is 21-hydroxylase, which is deficient in 90 per cent of cases. A lack of cortisol, due to the enzyme deficiency, leads to a compensatory hyperplasia of the adrenal gland, resulting in increased androgen production. In those with 46,XY chromosomes (and testes as gonads), increased androgen exposure does not alter the development of the reproductive organs from the ‘typical male’ pathway. For an individual with 46,XX chromosomes (and ovaries as gonads), increased levels of testosterone mean the development of the external genitalia occurs along a spectrum, ranging from ‘typical female’ to ‘typical male’ development. In these instances, CAH may be identified at birth with atypical genitalia (also referred to in medical literature as ambiguous genitalia). Late onset CAH, due to milder cases of enzyme deficiency, usually present later in childhood or in adolescence with precocious puberty or signs of hyperandrogenism.
Complete androgen insensitivity syndrome
Among the 46,XY intersex variations, one of the most common is complete androgen insensitivity syndrome (AIS). In people with AIS, the gonads are testes and produce hormones as per the ‘typical male’ pathway. However, the androgen receptors within the body are not able to respond to circulating androgens, thus, the androgens cannot influence development of the reproductive anatomy as it typically would. AMH produced by the testes still results in regression of the Müllerian ducts and the individual therefore has external female phenotype and male internal gonads (testes). At puberty, increasing levels of testosterone converts peripherally to oestrogen, resulting in female development of secondary sexual characteristics. Due to regression of the Müllerian ducts, there is no uterus or upper vagina, and individuals commonly present with primary amenorrhoea at puberty.
46,XY pure gonadal dysgenesis (Swyer syndrome)
Unlike in CAH or AIS, the gonad in Swyer syndrome does not develop into a testis or ovary; instead, a dysgenetic gonad develops. The gonad does not produce hormones or gametes. In the developing embryo, the lack of AMH and testosterone production leads down the ‘typical female’ pathway for internal and external development. The lack of hormone production from the gonad results in the absence of the onset of puberty.
Healthcare for people with intersex variations
The Australian Human Rights Commission is conducting a research project on ‘how best to protect the human rights of people born with variations in sex characteristics in the context of medical interventions’, which is still ongoing at the time of writing this article.5
Historically, healthcare for individuals with intersex variations was paternalistic and often surrounded by secrecy. Non-disclosure of the variation occurred and surgical and medical treatments were not always fully explained. This is no longer acceptable practice. Full disclosure of information and openness with patients and parents is considered vital.
Early (often in infancy) ‘normalising’ genital surgery for atypical genitalia has been the traditional practice of some medical professionals and still occurs in many centres today. This is a controversial topic and intersex advocates have been vocal in expressing their concern regarding non-medically necessary early surgery. In some parts of the world, there has been a move to delay surgery until the individual is old enough to be involved in the decision-making process.6 Promoters of early surgery believe that creating a more ‘normal’ genital appearance may promote parental bonding and reduce psychological distress for the child. Concerns regarding early surgery include potential alteration or damage to adult sexual function, with no good evidence that surgery improves long-term psychological outcomes.7 Irreversible cosmetic genital surgery may also restrict future choices for the small, but increasing, numbers of children with intersex variations choosing to reassign gender in later life.
People with intersex variations, and their parents or carers, may require or seek medical care for a number of reasons. This may include urgent situations such as salt-wasting crisis in a newborn with CAH. This risk must be rapidly identified and treated (with replacement steroids) to prevent death. Other clinical considerations for health professionals include: management of malignancy risk from the gonad; discussion about hormone replacement therapy; prevention of osteoporosis; fertility options; addressing concerns regarding sexual health or function; and offering appropriate psychological support and transitional care from paediatric to adult services. Holistic individualised care should be provided by health professionals with expertise in caring for people with variations of sex characteristics. Autonomy of the individual (including the child and future adult) should be respected, with ongoing support offered to optimise quality of life and wellbeing.
References
- Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Journal of Pediatric Urology 2006 Jun;2(3):148-62.
- Blackless M, Charuvastra A, Derryck A, et al. How sexually dimorphic are we? Review and synthesis. American Journal of Human Biology 2000 Mar;12(2):151-66.
- Carpenter M. The ‘normalisation’ of intersex bodies and ‘othering’ of intersex identities in Australia. Journal of Bioethical Inquiry 2018 May;7:1-9.
- Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. Journal of Pediatric Urology 2006 Jun;2(3):148-62.
- For further information visit: www.humanrights.gov.au.
- Pasterski V, Prentice P, Hughes IA. Consequences of the Chicago consensus on disorders of sex development (DSD): current practices in Europe. Archives of Disease in Childhood 2010 Aug;95(8):618-23.
- Kon AA. Ethical issues in decision-making for infants with disorders of sex development. Hormone and Metabolic Research 2015 May;47(5):340-3.
One Comment
Leave a Reply



For more stories and statistics…
https://interactadvocates.org/wp-content/uploads/2016/01/Intersex-Stories-Statistics-Australia.pdf