


The human genome can be described by considering its origin. For sexually reproducing species, the genome originates at the moment of conception; for humans that is when the 23 chromosomes from the egg cell of the mother combine with the 23 chromosomes from a sperm cell of the father. Together, those 46 chromosomes make up the human genome. Each chromosome is made from DNA, which is a polymer comprising nucleotides represented by the letters A, T, C and G. Our genome contains more than six billion of such letters, divided between the 46 chromosomes.The human genome can be described by considering its origin. For sexually reproducing species, the genome originates at the moment of conception; for humans that is when the 23 chromosomes from the egg cell of the mother combine with the 23 chromosomes from a sperm cell of the father. Together, those 46 chromosomes make up the human genome. Each chromosome is made from DNA, which is a polymer comprising nucleotides represented by the letters A, T, C and G. Our genome contains more than six billion of such letters, divided between the 46 chromosomes.
Our genome contains all the information required by life to program the single-celled embryo that is created at the moment of conception to divide in a perfectly coordinated fashion into hundreds, thousands and, eventually, billions of cells as the fetus develops in utero into a human being. Every tissue and structure in an individual’s body – such as blood cells, muscles, nervous system, heart, lungs, kidneys, bones – are all programmed and operated by the information in their genome. The remarkable precision of this program is dramatically illustrated in cases of identical twins, where it can run with almost complete reproducibility – not just eye and hair colour, but even the finest details of how our bodies develop, age and respond to disease and the environment; including, for example, where and when our hair greys, the complexion of our skin and where we accumulate fat and muscle.
Genomic variation
The information in our DNA makes us different. Virtually every disease, or our response to disease, is encoded in our genomes. Although the majority of the genome among humans is identical, of the six billion letters of information, approximately three million differ between two unrelated individuals. Among these millions of differences or ‘variants’, the majority are considered ‘benign’ because they have no damaging effect in the way our bodies work and are simply part of what creates the variation in our physical and biochemical make up. However, a small number of such variants can cause or predispose to disease. These ‘pathogenic’ variants may change the meaning of the genetic instructions by altering or removing letters in a segment directly translated by the cell, or a region that regulates gene activity. These changes can cause a cell or tissue to work poorly or not at all.
Genetic variation can involve a single DNA nucleotide or lead to the loss or rearrangement of huge chunks of DNA. Techniques such as karyotyping and fluorescent in-situ hybridisation have provided high-level resolution of large chromosomal duplications or rearrangements. Chromosomal microarrays have provided information about deletion and duplication of large segments of DNA, or copy number variants (CNVs). Genomic sequencing is now able to provide high-resolution information on small and large variants – including substitutions, deletions, insertions and complex rearrangements, such as inversions of large pieces of DNA.
We previously referred to benign and pathogenic variants as polymorphisms and mutations, but it has become clear over recent years that these lie on a continuum. Our understanding of the genome is still limited: there are many variants of unknown significance (VUS) that lie between our knowledge of benign and pathogenic. To complicate matters further, the severity or frequency of disease (so-called penetrance) that manifests from even well-characterised pathogenic variants can also be highly variable between individuals.1 This variable penetrance further confounds the interpretation of genomic information and limits the potential to predict disease from genomic information alone. Accordingly, accurate genomic interpretation is undertaken in the context of family history and patient presentation. Nevertheless, despite these complications, our existing knowledge of genomic information can already have powerful applications in clinical practice.
Revolutions in genetics
Our knowledge of the human genome builds on major leaps in the 19th and 20th centuries, particularly those of: defining the principles of heredity; resolving DNA’s double helix structure and the genetic code; and developing the first sequencing chemistries that could determine the order of nucleotides along a length of DNA. However, the last two decades have seen an unprecedented leap forward in information and insight into the human genome.
More than ten years ago, the ‘reference’ human genome sequence was published: a virtual tome that provided global infrastructure for biology.2 As a result, researchers around the world could access a common collection of genomic data and overlay information about DNA variation and function. Scientists could begin to compare DNA sequences between humans and other organisms to identify sequences common to all living things.
The original reference sequences lay the foundations for genetic exploration of the approximately 21 000 genes in the human genome, which act as the templates for producing proteins and other molecules. However, the smaller-than-expected number of genes hinted at the hidden complexity of the human genome and the regulatory mechanisms that affect the activity of genes. The observation that many of the conserved regions lay in areas between genes – sequences not directly involved in producing proteins – also suggested that protein-coding genes were not the only vital components of the genome.
Decoding the genome
International projects in the last ten years have attempted to add flesh to the bare bones of the genome sequence. The introduction of increasingly high-throughput technologies, such as microarrays and automated sequencing, provided a means to capture snapshots of genetic activity, which led to more detailed descriptions of which parts of the genome are used in different cells and at different times. These deeper explorations of the human genome led to a number of unexpected observations, such as that a single gene can be differentially spliced to encode multiple forms of a protein that have subtly different functions and the identification of genes that do not encode proteins at all, but rather function to produce other classes of regulatory molecules (referred to as noncoding RNAs).
As research into the activity of the genome has blossomed, so has research into the ‘epigenome’, which are tissue- and cell-type modifications to the biochemical environment of the DNA or direct chemical modifications to the DNA itself. These modifications, in turn, affect the activity of the genome and, ultimately, define how the genome behaves in different cells. Each new study answers more questions about how different cells in our body use the information encoded in the genome and how our characteristics are mediated by the environment we inhabit.3
As advances in sequencing technologies accelerated in the mid-2000s, the genomic data available to researchers grew from a trickle to a flood. These technologies were employed to create catalogues of variation in different human populations4 and thousands of people’s genomes from a cross-section of cultures have now been sequenced and released as global resources.5 6 Lower sequencing costs have also enabled researchers to ask health-related questions on a massive scale: some studies have catalogued mutations that drive a tumour’s growth by comparing the genomes of tumour and normal cells. Others have focused on mapping the functionality of significant proportions of the human genome or seeking DNA differences between people with and without complex diseases.
These projects have provided a far more nuanced view of genomic variation across the genome. Genetic variants across the genome may be inherited, induced by environmental factors, or arise from errors made while copying DNA for cell division.
Following a decade of rapidly declining costs of genomic sequencing, in parallel with increasingly sophisticated knowledge of how the genome works, genome sequencing is now making a transition from an almost exclusively research-based discovery tool to a routine clinic-based diagnostic tool.
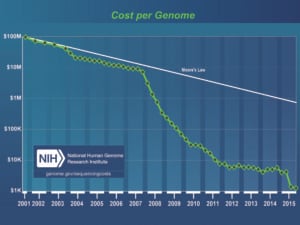
Figure 1. The cost of genome sequencing has declined at a hyper-exponential rate over the past decade, exceeding the rate of advance in semiconductor technology as predicted by Moore’s Law. Credit: Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) Available at: www.genome.gov/sequencingcosts. Accessed 04/2016.
Clinical implications of sequencing
The value of genome sequencing in the clinic – known as genomic medicine – is in using timely genetic diagnoses and better characterisation of disease to provide critical information to doctors and families. Information that predicts an individual’s risk of developing a disease, or their response to treatment, can optimise medical practice.Genomics is already affecting clinical practice through the diagnosis of heritable disease.7 Whole-genome sequencing (WGS) is transforming the diagnosis of thousands of rare, usually paediatric, diseases. Even for diseases that have not been described before, unbiased WGS can provide information that makes it possible to diagnose and define new genetic diseases.
Genomics is also starting to change oncology, by guiding the management and treatment of cancer. Cancer is ultimately a genetic disease: there are many ways that our genome can be mutated to cause cancer. Traditionally, cancer was treated according to its tissue of origin, but by understanding the genetic basis, treatment can be prescribed to target the specific molecular pathway individualised to that cancer.8 This ‘precision medicine’ approach attempts to apply the right drug in the right dose to the right patient at the right time.
Beyond diagnosis and guiding patient management, the most profoundly transformative application for genomics in healthcare is anticipated to be in disease risk stratification, early disease detection, and, ultimately, prevention of disease occurrence. The more we learn about what the information in our genomes means for health, the more accurately we can predict our health futures. As well as detecting disease predisposition, genomics also has the potential to anticipate adverse drug reactions, assess carrier status for recessive disease and predict the body’s response to different sorts of environmental exposures. The collective value of the health information derived from the genome is anticipated to eventually reach a tipping point where it will be advantageous for every individual to undergo genome sequencing and to incorporate this information as part of their health record.
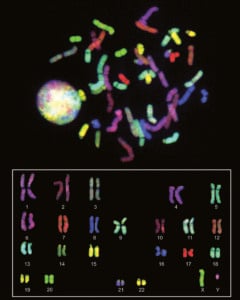
Figure 2. Cell nucleus and chromosomes stained by Spectral karyotyping (SKY). Although definitions vary according to the scientific circumstance, the human genome is typically defined as the full complement of genetic material in a cell. In cells other than germ cells (and mature red blood cells that lack most cellular organelles), genetic material is organised into 23 pairs of chromosomes inside the nucleus, and in the many copies of a small ring of DNA inside the cell’s energy-producing mitochondria.
The nuclear chromosomes are numbered in approximate order of size – from 1 to 22 – with the sex chromosomes (X&Y). One of each chromosome pair is inherited from an individual’s mother, and the other one from their father.
The future
By reading the information that forms the very essence of our bodies, genomics reveals the inner workings of our cells, in both health and disease. The power of sequencing derives from volume and the capacity to be able to compare newly sequenced genomes with many others to pinpoint the relevant variations. Genomic information will only gain interpretive power as it is combined with information about patients’ observable characteristics (their ‘phenomes’) and as more variants are understood and classified. Genomics is having an increasing impact in the clinic; and has the potential to reconfigure the nature of the interaction between the patient and the practitioner.

Figure 3. Chromosome organisation. Credit: Kate Patterson/Garvan. We each have about two metres of DNA crammed into the nuclei of our cells. The 3D structure of the human genome goes far beyond DNA’s double helix shape. When cells divide, the DNA is tightly wound around protein scaffolds into structures called chromosomes. The DNA is coiled systematically around histone proteins, packed into fibres and tied up into scaffolds with specific organisation. Throughout the cell cycle, threads of DNA coil and uncoil, depending on which pieces of genetic information are being used.
Mitochondrial DNA (mtDNA) sits outside the nucleus in a cell, and is inherited solely from the mother. Numbers of mitochondria in a cell vary, as do the numbers of copies of mitochondrial chromosomes, but each cell is thought to carry on average 1000 copies of the mtDNA, contributing around 0.1 per cent of total cellular DNA.
Recommended reading
Chen R, Shi L, Hakenberg J et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotech. 2016; advance online publication.
Feero WG, Guttmacher AE, Collins FS. Genomic medicine–an updated primer. New England Journal of Medicine. 2010; 362(21):2001-11.
Mattick JS, Dziadek MA, Terrill BN et al. The impact of genomics on the future of medicine and health. Medical Journal of Australia. 2014; 201(1):17-20.
PHG Foundation. Education: Genomics in mainstream medicine resources. Cambridge, UK: PHG Foundation; 2015 [cited 2016 26 April]. Available from: www.phgfoundation.org/education.
References
- Van Driest SL, Wells QS, Stallings S et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA. 2016; 315(1):47-57.
- Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004; 431(7011):931-45.
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489(7414):57-74.
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491(7422):56-65.
- Wheeler DA, Srinivasan M, Egholm M et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008; 452(7189):872-
- Schuster SC, Miller W, Ratan A et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010; 463(7283):943-7.
- McRae JF, Clayton S, Fitzgerald TW et al. Prevalence, phenotype and architecture of developmental disorders caused by de novo mutation. bioRxiv. 2016.
- Biankin AV, Waddell N, Kassahn KS et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012; 491(7424):
399-405.



Leave a Reply